
RASPA is a software package for simulating adsorption and diffusion of molecules in flexible nanoporous materials. The code implements the latest state-of-the-art algorithms for Molecular Dynamics and Monte Carlo in various ensembles including symplectic/measure-preserving integrators, Ewald summation, Configurational-Bias Monte Carlo, Continuous Fractional Component Monte Carlo, Reactive Monte Carlo, and Baker’s minimization. Applications of RASPA include computing coexistence properties, adsorption isotherms for single and multiple components, self- and collective diffusivities, reaction systems, and visualization.
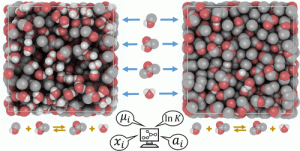
Brick-CFCMC is a new molecular simulation code for performing Monte Carlo simulations using state-of-the-art simulation techniques. The Continuous Fractional Component (CFC) method is implemented for simulations in the NVT/NPTensembles, the Gibbs Ensemble, the Grand-Canonical Ensemble, and the Reaction Ensemble. Molecule transfers are facilitated by the use of fractional molecules which significantly improve the efficiency of the simulations. With the CFC method, one can obtain phase equilibria and properties such as chemical potentials and partial molar enthalpies/volumes directly from a single simulation. It is possible to combine trial moves from different ensembles. This enables simulations of phase equilibria in a system where also a chemical reaction takes place.
Cassandra is an open source Monte Carlo package capable of simulating any number of molecules composed of rings, chains, or both. It can be used to simulate compounds such as small organic molecules, oligomers, aqueous solutions and ionic liquids. It handles a standard “Class I”-type force field having fixed bond lengths, harmonic bond angles and improper angles, a CHARMM or OPLS-style dihedral potential, a Lennard-Jones 12-6 potential and fixed partial charges. It does not treat flexible bond lengths. Fused ring systems should be simulated with caution. Cassandra uses OpenMP parallelization and comes with a number of scripts, utilities and examples to help with simulation setup.
Cassandra is capable of simulating systems in the following ensembles:
Canonical (NVT),
Isothermal-Isobaric (NPT),
Grand canonical (μVT), Constant volume Gibbs (NVT-Gibbs), and the
Constant Pressure Gibbs (NPT- Gibbs).
Towhee is a Monte Carlo molecular simulation code originally designed for the prediction of fluid phase equilibria using atom-based force fields and the Gibbs ensemble with particular attention paid to algorithms addressing molecule conformation sampling. The code has subsequently been extended to several ensembles, many different force fields, and solid (or at least porous) phases.
GOMC - GPU Optimized Monte Carlo is a parallel molecular simulation code. It is open-source software for simulating molecular systems using the Metropolis Monte Carlo algorithm. The software has been written in object oriented C++, and uses OpenMP and NVIDIA CUDA to allow for execution on multi-core CPU and GPU architectures. GOMC employs widely-used simulation file types (PDB, PSF, CHARMM-style parameter files).
GOMC can be used to study vapor–liquid equilibria, adsorption in porous materials, surfactant self-assembly, and condensed phase structure for complex molecules.
LAMMPS is a classical molecular dynamics (MD) code. As the name implies, it's designed to run well on parallel machines, but it also runs fine on single-processor desktop machines.
DL_POLY_4 general design provides scalable performance from a single processor workstation to a high performance parallel computer. It is supplied in source form under licence and can be compiled as a serial application code, using only a Fortran90 compiler, or as a parallel application code, provided an MPI2 instrumentation is available on the parallel machine. DL_POLY_4 offers fully parallel I/O as well as a netCDF alternative (HDF5 library dependence) to the default ASCII trajectory file.
GROMACS is a versatile package to perform molecular dynamics, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles.
It is primarily designed for biochemical molecules like proteins, lipids and nucleic acids that have a lot of complicated bonded interactions, but since GROMACS is extremely fast at calculating the nonbonded interactions (that usually dominate simulations) many groups are also using it for research on non-biological systems, e.g. polymers.
OpenMM is a toolkit for molecular simulation. It can be used either as a stand-alone application for running simulations, or as a library you call from your own code. It
provides a combination of extreme flexibility (through custom forces and integrators), openness, and high performance (especially on recent GPUs) that make it truly unique among simulation codes. biological systems, e.g. polymers. OpenMM includes everything one needs to run modern molecular simulations. It is extremely flexible with its custom functions, is open-source, and has high performance, especially on recent GPUs.
GULP is a program for performing a variety of types of simulation on materials using boundary conditions of 0-D (molecules and clusters), 1-D (polymers), 2-D (surfaces, slabs and grain boundaries), or 3-D (periodic solids). The focus of the code is on analytical solutions, through the use of lattice dynamics, where possible, rather than on molecular dynamics. A variety of force fields can be used within GULP spanning the shell model for ionic materials, molecular mechanics for organic systems, the embedded atom model for metals and the reactive REBO potential for hydrocarbons. Analytic derivatives are included up to at least second order for most force fields, and to third order for many.
PLUMED is an open-source, community-developed library that provides a wide range of different methods, which include:
enhanced-sampling algorithms,
free-energy methods, and
tools to analyze the vast amounts of data produced by molecular dynamics (MD) simulations.
These techniques can be used in combination with a large toolbox of collective variables that describe complex processes in physics, chemistry, material science, and biology.
PLUMED works together with some of the most popular MD engines, such as ACEMD, Amber, DL_POLY, GROMACS, LAMMPS, NAMD, OpenMM, DFTB+, ABIN, CP2K, i-PI, PINY-MD, and Quantum Espresso. In addition, PLUMED can be used to augment the capabilities of analysis tools such as VMD, HTMD, OpenPathSampling, and as a standalone utility to analyze pre-calculated MD trajectories.
PLUMED offers a large collection of collective variables that serve as descriptions of complex processes that occur during molecular dynamics simulations, for example angles, positions, distances, interaction energies, and total energy.
The Vienna Ab initio Simulation Package (VASP) is a computer program for atomic scale materials modelling, e.g. electronic structure calculations and quantum-mechanical molecular dynamics, from first principles.
VASP computes an approximate solution to the many-body Schrödinger equation, either within density functional theory (DFT), solving the Kohn-Sham equations, or within the Hartree-Fock (HF) approximation, solving the Roothaan equations. Hybrid functionals that mix the Hartree-Fock approach with density functional theory are implemented as well. Furthermore, Green’s functions methods (GW quasiparticles, and ACFDT-RPA) and many-body perturbation theory (2nd-order Møller-Plesset) are available in VASP.
In VASP, central quantities, like the one-electron orbitals, the electronic charge density, and the local potential are expressed in plane wave basis sets. The interactions between the electrons and ions are described using norm-conserving or ultrasoft pseudopotentials, or the projector-augmented-wave method.
To determine the electronic groundstate, VASP makes use of efficient iterative matrix diagonalisation techniques, like the residual minimisation method with direct inversion of the iterative subspace (RMM-DIIS) or blocked Davidson algorithms. These are coupled to highly efficient Broyden and Pulay density mixing schemes to speed up the self-consistency cycle. Note: VASP is commercial software and requires a license agreement with the University of Vienna.
CRYSTAL is a quantum chemistry ab initio program, designed primarily for calculations on crystals. It performs ab initio calculations of the ground state energy, energy gradient, electronic wave function and properties of periodic systems. Hartree-Fock or Kohn- Sham Hamiltonians (that adopt an Exchange-Correlation potential following the postulates of Density-Functional Theory) can be used. Systems periodic in 0 (molecules, 0D), 1 (polymers, 1D), 2 (slabs, 2D), and 3 dimensions (crystals, 3D) are treated on an equal footing. The program can automatically handle space symmetry: 230 space groups, 80 layer groups, 99 rod groups, 45 point groups are available. In the case of polymers it can treat helical structures (translation followed by a rotation around the periodic axis). Note: Crystal is commercial software.
CP2K is a quantum chemistry and solid state physics software package that can perform atomistic simulations of solid state, liquid, molecular, periodic, material, crystal, and biological systems. CP2K provides a general framework for different modeling methods such as DFT using the mixed Gaussian and plane waves approaches GPW and GAPW. Supported theory levels include DFTB, LDA, GGA, MP2, RPA, semi-empirical methods (AM1, PM3, PM6, RM1, MNDO, …), and classical force fields (AMBER, CHARMM, …). CP2K can do simulations of molecular dynamics, metadynamics, Monte Carlo, Ehrenfest dynamics, vibrational analysis, core level spectroscopy, energy minimization, and transition state optimization using NEB or dimer method. CP2K is written in Fortran 2008 and can be run efficiently in parallel using a combination of multi-threading, MPI, and CUDA. It is freely available under the GPL license.